
The development of biosimilars for dermatological and other indications has been an area of active development. Andrew Blauvelt, President of the Oregon Medical Research Center, Portland, Oregon, USA and past chair of Biosimilars working group for National Psoriasis Council, provided a review of key concepts and recent developments of biosimilars in dermatology. The US Food and Drug Administration (FDA) defines a biosimilar as a biological product that is highly similar to a reference product, notwithstanding minor differences in clinically inactive components, and with no clinically meaningful differences in terms of safety, purity, and potency. However, because biosimilars are created using different cell lines and different culture systems, they are not 100% identical to their parent compounds, and so cannot be regarded as bio-generics. As with generic drugs, the aim of developing biosimilars is to reduce the cost and increase access to biologic drugs for those patients who need them most. The promise of cost reduction has not yet been fully realized, however. While several biosimilars have been approved, few have reached the market due to efforts of the manufacturers of originator drugs to extend patent expiration.
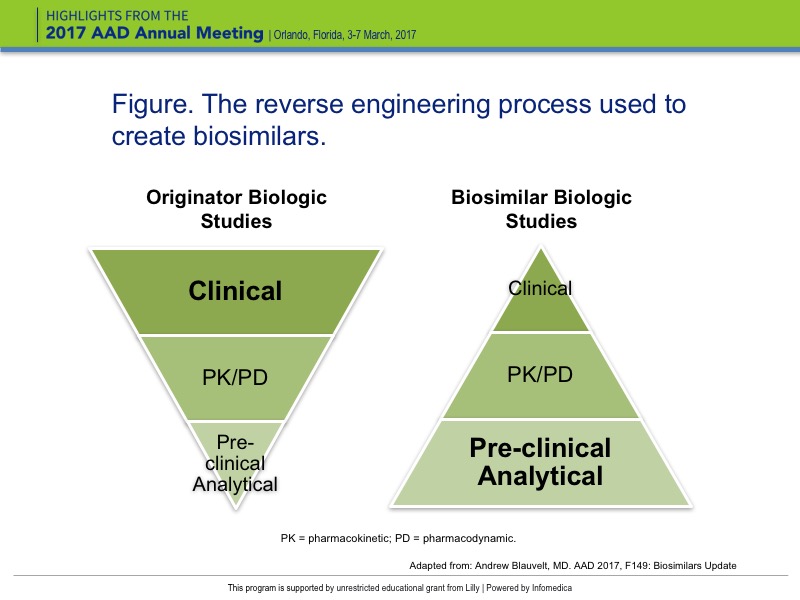
The clinical development models for originator biologics and biosimilars are strikingly different. For originator biologics, the investment in preclinical and analytical research in in vitro and mouse models is relatively small compared to clinical trials; particularly phase 3 trials, which can enroll thousands of patients. The total cost of developing an originator biological drug is estimated at $1B to $2.25B. In contrast, the lion’s share of investment for biosimilars involves preclinical analytical studies comparing the originator molecule with the biosimilar with regard to target-binding affinity, glycosylation patterns, additives, and other parameters. In addition, the requirement for approval is limited to a relatively small, single study. As a result, the estimated cost to develop a biosimilar is $250 million – about one-quarter to one-fifth that of developing original biologic.
In the reverse engineering process used to create biosimilars, the known protein structure and amino acid sequence of the originator molecule is used as a starting point. By working backwards, the DNA sequence required to code for that amino acid sequence is identified (Figure).
{kind=link}
The process of creating a biosimilar biologic medical product is complex. Biologics are produced in cell culture. First, the cell culture is modified to produce recombinant proteins. The cell line is grown in a controlled environment, and the recombinant drug is extracted, purified, and formulated into a finished product. The goal is to create a biosimilar with a similar fingerprint as the originator drug, in terms of primary structure, impurities, biological structure, high order structure, and posttranslational modifications, which involve changes that occur to the protein when it’s secreted from the cell.
Integration of data from multiple analytical and functional side-by-side tests against the original molecule provides the best view of biosimilarity.
Manufacturing changes over time in an originator molecule may lead to divergence; that is, changes in the nature of a biologic drug over time. Post-marketing comparative biosimilarity is not required, which is a reason why some regulators do not endorse interchangeability of biosimilars and originator drugs.
Another area of uncertainty is the potential for multiple biosimilars to be approved based on the same originator drugs. These biosimilars are required to be compared with the originator drug but not with each other, which may raise questions about whether there are significant differences among the biosimilars.
The FDA requires a minimum of one comparator equivalence assessment trial for approval, and the biosimilar does not need to be studied for all potential indications. If the drug is deemed “highly similar”, demonstrates equivalence in one key indication, and the mechanism of action is the same across disease states, the FDA may use the extrapolation of data from one indication to another to provide approval. Note, however, that equivalence trials of TNF blocker biosimilars are mostly being done in psoriasis.
In an equivalence study, parameters for how equivalent the drug needs to be are set up prior to the study. To demonstrate equivalence, the mean response and the measurement error on assessment of clinical response has to be between these “goal posts.” If results are outside the prespecified margin of equivalence, the biosimilar will not be approved. Equivalence studies are typically simple in design, with no placebo control, and a single switch. An example is the ABP 501 trial used to support approval of Amgen’s biosimilar adalimumab.
A biosimilar may be designed as “interchangeable”, with the originator compound, if 1) it has demonstrated that it can be expected to produce the same clinical result as the reference product in any given patient, and if 2) it has demonstrated that there is no risk in terms of safety or diminished efficacy in switching back and forth between the biosimilar and the reference product. This designation requires data from an interchangeability trial in which patients are switched between the biosimilar and the reference product at least 3 times. The Sandoz biosimilar etanercept is a designated interchangeable biosimilar.
Clinically, some of the implications associated with the use of biosimilars remain to be seen. There are concerns regarding extrapolation, the potential impact of differences in the patient populations across indications, and the lack of FDA guidance on pharmacovigilance specific to biosimilars.
The ability of pharmacists to switch from a reference product to a biosimilar with the prescribing physician’s approval may differ from state to state.